7 转录组拼接质量检查[Transcriptome Assembly Quality Assessment]
对于拼接得到的转录组,通过三种方法进行拼接质量评价。首先检查拼接转录本对原始数据的代表能力,其次分析拼接结果中完整转录本的比例,最后检查保守的直系同源基因评价组装的完整性。
For the transcriptome, three methods were used to evaluate the splicing quality. First check the transcripts’ representation of the original data, then analyze the proportion of complete transcripts in the splicing results, and finally check the conserved ortholog content to assess the integrity of the assembly.
7.1 原始reads比对至拼接转录本[Align reads back to assembled Transcript]
理想情况下,拼接转录组应该代表至少约80%的输入RNA-Seq Reads。 其余未组装的读段可能对应于表达水平较低的转录本,其覆盖范围不足以实现组装,或者是质量较低或异常的Reads。
Ideally, at least ~80% of your input RNA-Seq reads are represented by your transcriptome assembly. The remaining unassembled reads likely corresponds to lowly expressed transcripts with insufficient coverage to enable assembly, or are low quality or aberrant reads.
方法[Method]
使用Bowtie2(v2.3.5)将测序所得的原始reads比对至转录本以评估当前组装转录本对测序数据的代表能力。
Bowtie2 (v2.3.5) was used to compare the original reads obtained from sequencing to the transcript to examine the RNA-Seq read representation of the assembly.
总reads序列数[total reads]: 55010431
总计比对率[overall alignment rate]: 91%
双端正常比对[paired reads concordantly aligned]: 51%
双端异常比对[paired reads aligned discordantly]: 8%
剩余序列单端模式比对[other reads aligned by single end mode]: 31%
注:
- 该步骤以bowtie2工具进行比对。比对所用的组装转录本序列为各基因对应的isoform序列。
- 双端异常比对包括:比对方向与pair-end测序的方向不一致;read1和read2的插入片段长度过长。
- 此处需要考虑所有能比对上的序列,包括配对和未配对的比对。典型的转录组拼接能够将绝大多数的所有读取映射回组装,且匹配到的映射片段中70-80%应为成对匹配。
- 对于未能成对比对的序列,其原因可能是由于:转录本较短或因可变剪接等原因导致对应isoform仅包含reads的一端。
- 其余未装配的reads可能对应于低表达转录本,覆盖度不足而无法被组装,也可能为低质量或异常reads。
比对结果总结[align summary] : align_stats
详细比对结果[align detials] : bowtie2.alignBack.bam
7.2 保守基因检测[Assemble]
为了评价拼接结果的质量,评估真实转录本被拼接过程成功重构的比例,以及重构的完整程度。借助BUSCO工具,分析近缘物种中保守基因拼接的完整性和准确性。BUSCO 软件根据OrthoDB 数据库,构建了几个大的进化分支的单拷贝基因集。将转录本拼接结果与该基因集进行比较,根据比对上的比例、完整性,来评价拼接结果的准确性和完整性。
方法[Method]
挑选每个基因中的最长转录本(避免不同isoform被错误计算为多拷贝),使用BUSCO(v4.0.1)进行保守基因检测。物种参数选择–auto-lineage-euk,交由BUSCO在真核生物范围内自动进行选择。
Pick the longest transcript in each gene (to avoid different isoforms from being miscalculated as multiple copies) and use BUSCO (v4.0.1) for conservative gene detection. Species parameter selection –auto-lineage-euk, which will be selected automatically by BUSCO within the scope of eukaryotes.
保守基因检测结果
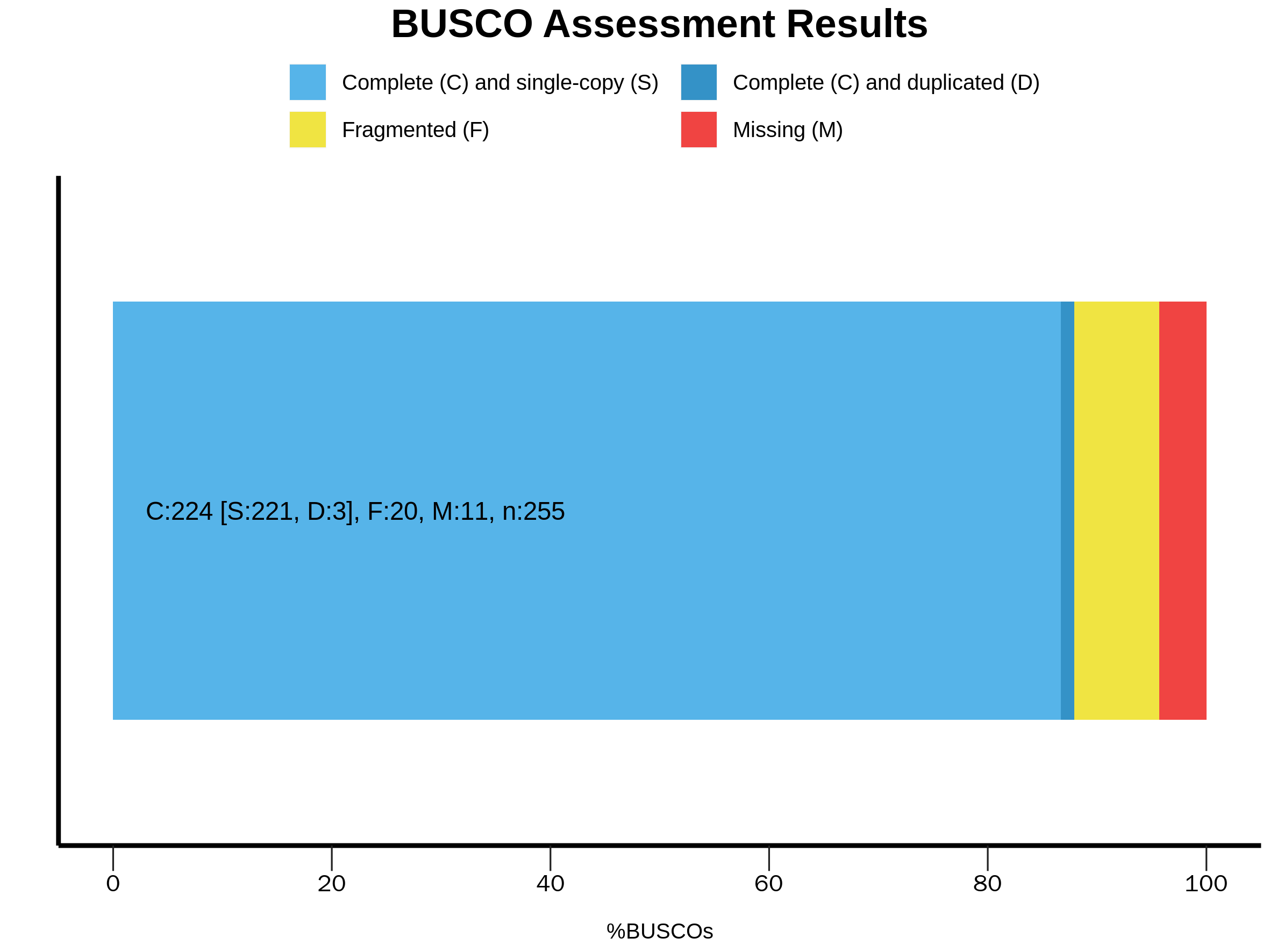
注:
* C (Complete) 拼接转录本覆盖到的BUSCO测试基因
* S (single-copy) 单拷贝基因
* D (duplicated) 多拷贝基因
* F (fragment) 部分比对基因
* M (miss) 未比对到的基因
* Total 测试所用保守基因总数
比对所用数据库等详细信息参见:short_summary
7.3 全长转录本估计[Assemble]
评估转录组装配质量的指标之一是检查全长或接近全长的转录本数量。该指标表征拼接所得转录本完整性。 在缺少参考基因组/转录组的情况下,将组装的转录本与所有已知蛋白质进行比对,并统计最优匹配的覆盖度达到X%的转录本数量。
One metric for evaluating the quality of a transcriptome assembly is to examine the number of transcripts that were assembled that appear to be full-length or nearly full-length. In the absence of a reference genome/transcriptome, a more general analysis to perform is to align the assembled transcripts against all known proteins and to determine the number of unique top matching proteins that align across more than X% of its length.
方法[Method]
使用BLAST+(v2.9.0)进行比对,以SwissProt数据库进行建库。对数据库中的每条序列仅保留最佳匹配的转录本。对于一条转录本与一条数据库蛋白有多个不连续比对(包含多个高得分片段对的BLAST匹配,HSPs)的情况。对多个HSPs进行分组,并共同计算覆盖度。 Use BLAST + (v2.9.0) for comparison, and SwissProt database. Only the single best matching Trinity transcript is reported for each top matching database entry.It is sometimes the case that a single transcript will align to a single protein sequence with several discontinuous alignments (a BLAST hit containing multiple high scoring segment pairs (HSPs)). First group those multiple HSPs per transcript and database hit, and compute the alignment coverage based on the grouped HSPs.
比对结果统计
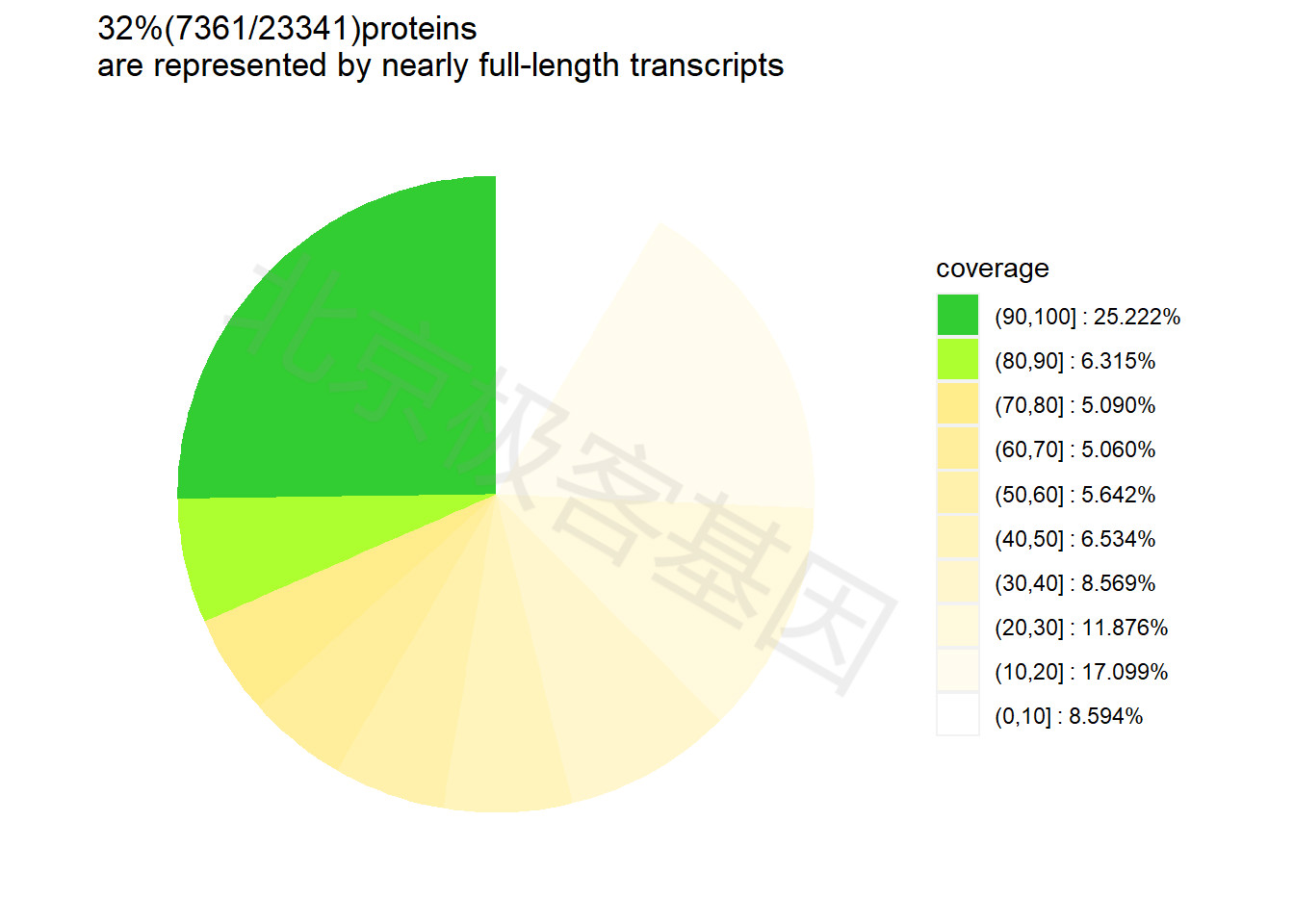
具有大于80%转录覆盖度的转录本一般可以认为是接近全长转录本。
全长转录本占比较低与测序数据量、数据质量以及基因组特征有关。
该步骤使用的蛋白质数据库为 SwissProt 。
拼接转录本对SwissProt数据库比对结果 [Assembled transcripts blastx to UniPort SwissProt] : blastx2uniport.format6
