1 项目介绍[Project Introduction]
无参转录组分析主要用于在缺少参考基因组的情况下,获取样本中的转录组数据,用于样本、物种之间在转录本类型和转录水平的比较,进而挖掘这些转录本之后的生物学意义。
目前我们已经能够获取到大量物种的参考基因组,但相对于自然界的万千物种而言,参考基因组数据仍然不足,例如具有重要生态和演化意义的非模型生物、癌症样本、微生物组样本。由于基因组通常具有较大的体积且存在大量重复序列,目前而言,对基因组进行测序和组装仍然是非常昂贵的。
RNA-seq技术能够用于研究转录组的各个方面。其应用包括基因发现,可变剪接检测,差异表达分析,融合检测和SNP等变体的鉴定以及转录后编辑。 RNA-seq相对于诸如微阵列等较早的技术,优势之一是无需参考基因组和注释即可生成和分析数据,因此它可以对非模型生物进行转录组范围的分析。当没有参考基因组可用时,转录组直接从RNA-seq读数重新组装。转录组作为基因组总体序列的一小部分,RNA测序能够以较低成本为下游的分析和应用提供参考转录组。
1.1 无参转录组的主要应用[Main applications of nonparticipating transcriptome]
由于实验简单、价格低廉等因素,无参转录组分析获得了广泛的应用。无参转录组的主要应用包括:
- 转录本水平研究
经过拼接获取样本中的转录组,捕获样本中基因的表达水平。并结合功能注释,为分子标记开发、物种演化研究等提供基础。
- 差异转录组
研究同一物种表型有差异的mRNA序列,挖掘与表型相关的差异转录本结构。
- 比较转录组
在演化研究中,通常会对基因组、转录组进行比较。但对于大多数生物类群,仅有较少的代表性物种具有基因组数据。通过无参拼接得到参考转录组,为此提供了技术基础。已被广泛应用于研究近缘物种间的亲缘关系以及不同物种或亚种间mRNA序列差异,挖掘明显受到正向选择或负向选择的基因等。
- 辅助基因注释
若仅基于基因组测序数据,现有的基因预测软件很难准确地预测基因,借助转录组拼接可以促进复杂基因结构的预测,提高其准确性。
1.2 无参转录组的分析流程[De novo transcriptome assembly and analysis workflow]
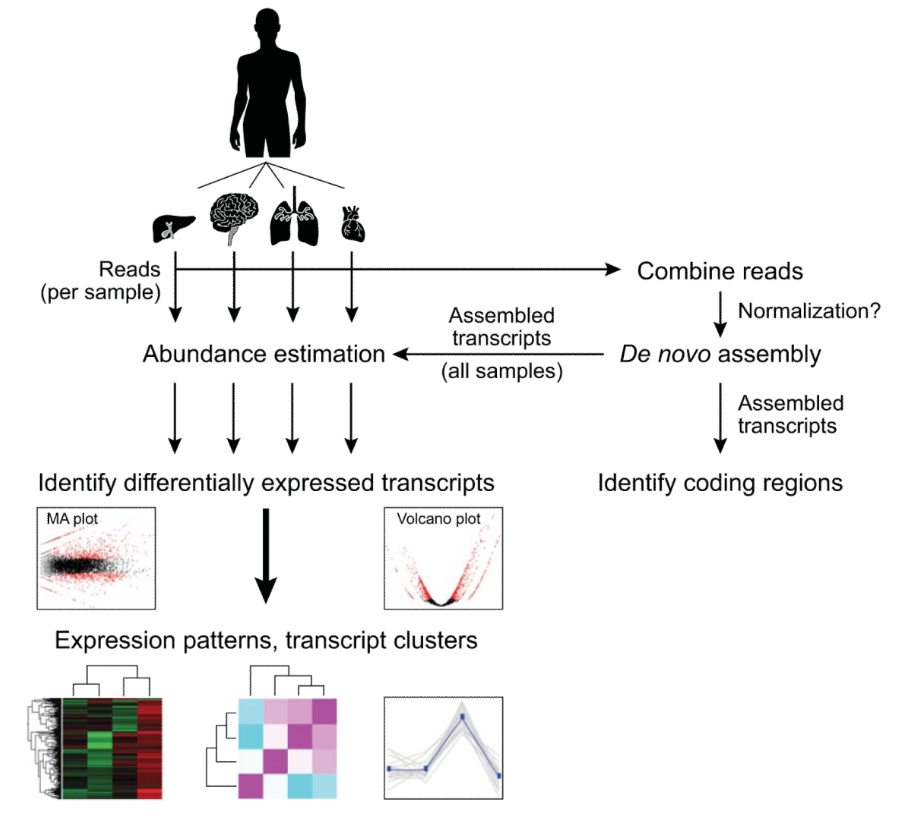
常规组学分析中,一般依赖于基因组的序列和注释信息。无参分析经过拼接注释等过程获取参考转录本序列和注释,从而实现对参考基因组的替代。无参转录组的分析流程主要包括:转录本拼接、转录本注释、表达定量和下游分析。
测序所得的Reads数据是一些长度在几百bp的的短序列,具体长度取决于建库测序策略。在对原始测序进行质量检查和过滤后,依据这些较短的序列中的信息,经过拼接延伸聚类等过程,得到转录本序列。因为转录过程的复杂加工,在得到的拼接结果中,同一个基因可能对应多个转录本,且常会包含大量的非编码RNA、rRNA等。通常得到转录本序列之后,对这些转录本序列进行质控,以检测拼接过程的效果和可能存在的问题。常用的质控方法包括检查保守基因的完整度、和蛋白数据库进行比对评价拼接完整性、检查原始测序数据的回帖比率等。
经过拼接,我们能够得到转录本的序列信息,但这些转录本是否编码蛋白、编码的蛋白序列和功能都还未知,因而需要进行转录本的注释,包括开放阅读框(ORF)预测和功能注释。这一步的注释依赖于来自其它物种的蛋白功能、结构域注释数据,这些数据一般来自UniPort、Pfam等公共数据库。至此,我们就得到了目标物种的转录组序列和注释信息,可用于在其他下游分析中充当参考基因组。
基于拼接所得的参考转录组,我们能够进行基于表达量的样本比较、获取差异表达基因并进行功能概括。同时也能够进行变异分析(SNP、InDel)、鉴定简单重复序列(SRR)用于开发分子标记,以及用于比较转录组分析等其他分析技术。